A Composable and Modular Framework for Protein Structure Prediction on HPC
DOI: https://doi.org/10.1145/3785462.3815886
PEARC '26: Practice and Experience in Advanced Research Computing, Minneapolis, MN, USA, July 2026
Current AI protein structure prediction models involve multi-stage processing that combines deep neural networks with bioinformatics tools such as multiple sequence alignment (MSA). Researchers increasingly rely on intermediate or penultimate-layer activations from these models for downstream tasks including contact prediction, binding-site identification, and model interpretability. We describe the VizFold plugin, a modular framework that can be extended toward end-to-end composable pipelines. We demonstrate feasibility through standardized hook-based tracing for ESMFold and Boltz-2, archive validation, and reproducible deployment on an HPC cluster using managed caches, modules, quotas, and Slurm workflows. The framework extracts attention maps from user-selected layers and exports intermediate representations in a backend-specific run bundle (Boltz) or a canonical archive tree (ESMFold), with shared trace text conventions and explicit provenance metadata suitable for cross-model comparison. We provide step-by-step documentation for instrumentation and deployment so that other groups can reproduce or extend the pipeline on their own clusters. The framework and instrumentation code are open-source and available at https://github.com/AI2Science/vizfold-foundation.
ACM Reference Format:
Jayanth Vennamreddy, Arish Virani, Kevin Yin, Vishal Ramasubramanian, Jeeva Ramasamy, Giri Krishnan, and Suresh Marru. 2026. A Composable and Modular Framework for Protein Structure Prediction on HPC. In Practice and Experience in Advanced Research Computing (PEARC '26), July 26--30, 2026, Minneapolis, MN, USA. ACM, New York, NY, USA 4 Pages. https://doi.org/10.1145/3785462.3815886
1 Introduction and Background
AI-based methods have transformed protein structure prediction. AlphaFold2 [1, 2] demonstrated near-experimental accuracy and was recognized with the Nobel Prize; since then, newer architectures have expanded the design space, including AlphaFold3 [3], Boltz-2 [4], and ESMFold [5].
Despite their diversity, these models share many similarities and often include three components: (1) input encoding—MSA-based co-variation (AlphaFold2/3, Boltz-2) or protein language model embeddings (ESMFold via ESM-2 [6]); (2) a multi-layer attention network (Evoformer or Pairformer) that builds pair representations; and (3) a structure module that converts these into 3D coordinates via prediction or diffusion. AlphaFold and Boltz architectures execute these stages sequentially. Accessing intermediate outputs at each stage has also become critical for a wide range of downstream applications and interpretability research. Yet constructing flexible, reproducible, and modifiable workflows involving these components remains challenging because implementations differ in how inputs and outputs are represented and exposed through APIs. Thus, a modular and composable framework for computing these components on HPC systems can support a wide range of applications in protein science.
In this work, we present an initial step toward a fully modular multi-backend framework for protein structure prediction. We demonstrate these capabilities with ESMFold and Boltz-2 backends. In §2 we elaborate the broader context for the long-term vision; §3–8 present the interface, methods, engineering challenges, HPC deployment, and evaluation.
2 Vision and Scope
The long-term goal is a system in which users can compose predictors, MSA generation, and post-hoc analysis stages, with intermediates cached on cluster scratch and reused across Slurm job steps. This paper demonstrates a concrete subset: (i) hook-based tracing with distinct on-disk layouts—a canonical archive tree for ESMFold versus a flat Boltz-2 run bundle (pred/, attn_txt/, act_npz/, arc_png/, manifest.json)—using shared trace-text conventions and explicit provenance metadata; (ii) environment patterns on Georgia Tech PACE ICE; (iii) validation of extracted artifacts; and (iv) representative end-to-end runs including arc-diagram exports. Benchmarking, accuracy claims, and workflow-manager support are future work.
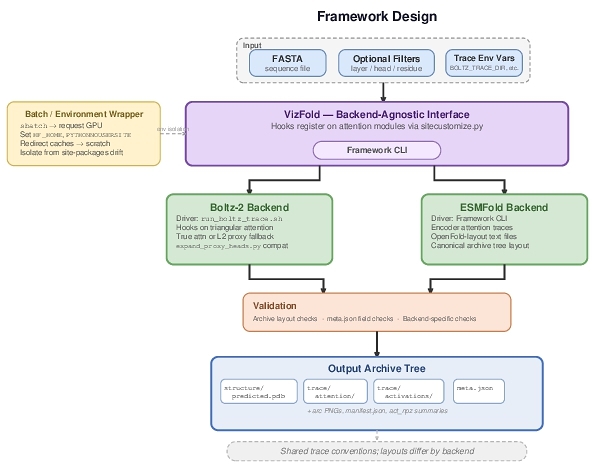
3 Framework Design
The VizFold plugin defines a backend-agnostic interface with FASTA input, optional layer/head/residue filters, and validated intermediate artifacts. ESMFold writes a canonical archive tree (structure/, trace/attention/, trace/activations/, meta.json). Boltz-2 currently writes a flat run directory (pred/, attn_txt/, act_npz/, arc_png/, manifest.json) with component-separated traces under attn_txt/components/...; strict validation enforces internal consistency of paths and metadata. Hooks do not alter weights. Batch wrappers isolate the environment (HF_HOME, PYTHONNOUSERSITE, etc.); a researcher requests a GPU via sbatch, sets trace variables, and invokes the backend driver. Figure 1 shows the architecture.
4 Modular Components of AI Protein Structure Prediction
4.1 Computing MSA
For MSA-based models (AlphaFold2/3, Boltz-2), the first stage aligns the input sequence against a protein database. The VizFold plugin does not re-implement MSA generation; instead, it consumes the MSA produced by the backend (or operates without one in single-sequence mode) and extracts attention and pair representations from subsequent stages.
4.2 Using Embeddings from a Protein Language Model
ESMFold replaces MSA with ESM-2 [6], a protein language model trained on ∼ 250M sequences. Our hooks capture attention maps and hidden states from all 36 encoder layers, plus IPA attention and backbone positions from the structure module at each recycling step.
4.3 Extracting Intermediate Representations
Hooks capture intermediate tensors dynamically without modifying the underlying model weights.
ESMFold. We hook EsmForProteinFolding [7] at each encoder self-attention layer, forcing output_attentions=True. Attention maps are (N + 2)2 before slicing to N × N. msa_row files contain reformatted encoder self-attention for arc-diagram compatibility [8]. Structure-module extraction is detailed in §4.4.
Boltz. Setting BOLTZ_TRACE_DIR activates a sitecustomize.py import hook on triangular attention modules [4, 9]. When true attention tensors are exposed through the attention modules, we export head-structured matrices; otherwise we fall back to an L2 norm over pair channels of the triangular-attention output tensor, normalized to [0,1]. This proxy is not an attention probability distribution. It is a visualization/diagnostics surrogate recorded as proxy_pair_norm (or related) in component_status.json. When BOLTZ_TRACE_HEAD=all but only a single proxy head is emitted, we optionally duplicate head blocks via expand_proxy_heads.py to preserve tooling expectations (duplicate heads are identical copies, not independent attentions). Outputs include VizFold-compatible msa_row and triangle trace text under attn_txt/, optional act_npz/ summaries, arc_png/ diagrams, and manifest.json. An experimental MSA hook (BOLTZ_TRACE_EXPERIMENTAL_MSA=1) may write additional traces when compatible modules are discoverable.
4.4 Extracting from the Structure Module
In ESMFold, the IPA-based structure module iteratively refines backbone frames over eight recycling steps; our CapturingSoftmax wrapper captures per-block attention ([B, 12, N, N]) and backbone positions ([8, 1, N, 14, 3]) at each step. Boltz-2 uses a diffusion-based structure module; Pairformer pair representations (from the triangular-attention hooks above) serve as the closest analogue. Per-diffusion-step coordinate extraction is deferred to future work.
5 Issues Faced by Extraction and Solutions
ESMFold. EsmAttention discards raw weights; we hook EsmSelfAttention directly and inject output_attentions=True, inspecting the call signature at runtime to handle positional-vs-keyword mismatches across transformers builds. The IPA softmax requires wrapping in an nn.Module node, so we wrap it in a CapturingSoftmax subclass [10]. Full-layer trace archives can reach 6–15 GB; the --layers and --heads CLI flags select subsets, and .detach() prevents gradient-graph memory inflation.
Boltz. Triangular-attention internals vary across releases; we attempt true-attention capture first and fall back to a pair-norm proxy (L2 over pair channels, normalized to [0,1]), recording provenance in component_status.json. expand_proxy_heads.py duplicates the single proxy head into multi-head slots for tooling compatibility. Tracing activates only when BOLTZ_TRACE_DIR is set; otherwise Boltz runs unmodified.
6 HPC Deployment Challenges and Solutions
6.1 ESMFold
The 8.4 GB checkpoint can exceed home-directory quota on PACE ICE; we redirect HF_HOME to scratch and pin transformers==4.38.2 with PYTHONNOUSERSITE=1. Weights load via use_safetensors=True to avoid the pickle path affected by CVE-2025-32434 [11].
| Component | Version |
|---|---|
| Python | 3.10 |
| PyTorch | 2.10 |
| transformers | 4.38.2 |
| CUDA | 12.4 |
| GPU | NVIDIA H200 |
| Cluster | PACE ICE |
| Output | Shape | Source |
|---|---|---|
| Attention (36) | [1, 40, N, N] | Encoder |
| Activations (36) | [1, N + 2, 2560] | Encoder |
| $s\_s$ | [N, 1024] | Structure |
| IPA (8 blocks) | [1, 12, N, N] | Structure |
| Backbone | [8, 1, N, 14, 3] | Structure |
| PDB | — | Full model |
6.2 Boltz
Boltz requires compiled C++ extensions and RDKit [12]; under mixed conda/pip environments, RDKit can fail on compiled symbols at inference time. Installing Boltz extras with pip install --no-deps while keeping PyTorch and CUDA conda-pinned avoids conflicts. Our Slurm driver run_boltz_trace.sh redirects caches to scratch (TMPDIR, PIP_CACHE_DIR, XDG_CACHE_HOME, HF_HOME, TORCH_HOME, WANDB_DIR). One-time environment creation (setup_boltz_env.sh) exports PYTHONNOUSERSITE=1 (and PIP_USER=0) during that script so pip installs avoid user site-packages; we recommend the same in interactive or Slurm shells when diagnosing mixed-path import issues. The --no_kernels flag disables runtime CUDA compilation on ICE H100 nodes.
7 Benefits of HPC Execution Environments
ESMFold's checkpoint (∼ 8.4 GB) plus dense traces (up to ∼ 2 GB per run) exceed typical workstation resources. An H200 node reduced a 191-residue run to under 30 s. On one H100, our default Boltz-2 trace (layer 0, single residue, top-k = 50, BOLTZ_TRACE_HEAD=all with proxy head expansion, including validation and arc rendering) completed in 204 s (Slurm Elapsed), while a deeper demonstration (Pairformer layers {0, 8, 16, 24, 32} at residues {18, 50}, top-k = 50) completed in ∼ 272 s (∼ 4.5 min) end-to-end. HPC also enables cross-model workflows—running the same target through multiple backends and caching intermediates on scratch for comparison—that are impractical locally.
8 Results and Evaluation
8.1 ESMFold
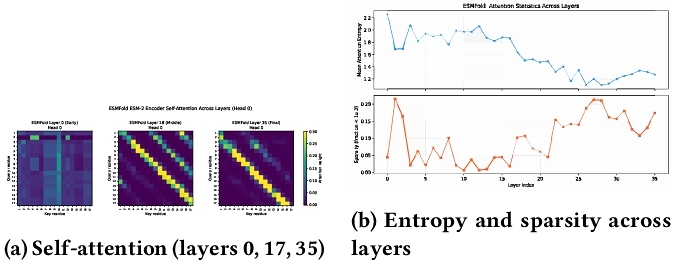
A 17-residue CPU run yielded 36 attention tensors [1,40,17,17], 36 activations [1,19,2560], out.s_s [17,1024], and reference-format text files [8]. Table 2 summarizes shapes. Figure 2 shows attention evolution and per-layer statistics. 6KWC (191 residues) on H200 ran in < 30 s including load; synthetic checks validated IPA and backbone tensors. These layer-wise entropy and sparsity statistics themselves represent a downstream analysis enabled by the extracted archives, illustrating how stored intermediates support interpretability without re-running inference.
8.2 Boltz
Unless noted otherwise, our default ICE/Slurm run configuration traces layer 0 at residue 18, producing three trace text files and twelve arc diagrams after optional proxy head expansion. Separately, we also demonstrate deeper Pairformer tracing by selecting multiple layers. On PACE ICE (one H100), tracing layers {0, 8, 16, 24, 32} with top-k = 50 took ∼ 4.5 min end-to-end. The run used an empty MSA (single-sequence mode) and, for layers {0, 8, 16, 24, 32} at residues {18, 50} with top-k = 50, produced 25 top-level trace text files and 100 arc-diagram PNGs (4 heads after proxy expansion), plus manifest.json; component_status.json recorded proxy_pair_norm. Figure 3 shows edge patterns evolving across Pairformer depth: early layers are diffuse while deeper layers concentrate on fewer residue pairs, consistent with iterative geometric refinement. ESMFold inference is deterministic; repeated runs produce identical archives. Boltz traces with a fixed seed (--seed 0) produced consistent Pairformer proxy traces across runs; diffusion-stage coordinates may vary. These visualizations demonstrate a second downstream use case: cross-layer structural analysis from archived traces.
8.3 Performance Overhead and Downstream Impact
To quantify the extraction overhead on HPC workloads, we compared vanilla inference against hooked tracing. Boltz-2 exhibited ${\sim }5\%$ wall-time overhead for hooked versus vanilla boltz predict under our default targeted-layer trace settings (predict step only; excludes arc rendering and post-run validation). ESMFold hook overhead was under $5\%$ for targeted layer extraction (attention-only); the dominant cost is GPU-to-CPU transfer of captured tensors, not additional computation. Full 36-layer tracing adds further I/O overhead from writing the resulting ∼ 2 GB trace archives. Despite this cost, the exported attention and pair summaries enable downstream analytical tasks without repeating full-model inference—for example, lightweight classifiers for binding-site prediction or layer-wise entropy metrics for flagging potentially misfolded regions.

9 Conclusion
We described the VizFold plugin framework and its hook-based extraction mechanisms for ESMFold and Boltz-2 on an HPC system (Georgia Tech PACE ICE). ESMFold conforms to a canonical archive tree (structure/, trace/, meta.json), while Boltz-2 currently emits a flat run bundle (pred/, attn_txt/, act_npz/, arc_png/, manifest.json) with explicit provenance in component_status.json. Unifying these into one on-disk standard is straightforward engineering (export/adapt) and is the next integration step for cross-backend dashboards. The intermediates from this plugin can be extended for downstream tasks—binding-site prediction from attention patterns, misfolded-region detection via layer-wise entropy, or lightweight classifiers on pair features. Our work can be extended to cross-model comparison on shared targets and broader backend support including OpenFold variants, together with adapters or export steps that map Boltz-2’s flat run bundle onto the same dashboards as ESMFold's canonical tree.
Data and Code Availability
The VizFold plugin framework, extraction hooks, and deployment scripts are open-source. The code and documentation can be accessed at https://github.com/AI2Science/vizfold-foundation.
Acknowledgments
Generative AI tools assisted in drafting portions of this manuscript from author-provided technical materials; all AI-generated text was reviewed and verified by the authors. This work is supported by the National Science Foundation under Award No. 2502793. Additional support was provided by Georgia Institute of Technology and the PACE ICE cluster.
References
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli, and D. Hassabis, “Highly accurate protein structure prediction with AlphaFold,” Nature, vol. 596, no. 7873, pp. 583–589, 2021.
- Gustaf Ahdritz, Nazim Bouatta, Sachin Kadyan, Qinghui Xia, William Gerecke, Timothy J. O'Donnell, Daniel Berenberg, Ian Fisk, Niccolò Zanichelli, Bo Zhang, Arber Nowber, Buxin Cormier, Hannes Jin, Lucy Wang, Parmida Banerjee, Bradley L. Pentelute, Dima Kozakov, Daphne Vogel, Teresa Head-Gordon, and Mohammed AlQuraishi. 2024. OpenFold: Retraining AlphaFold2 yields new insights into its learning mechanisms and capacity for generalization. Nature Methods 21 (2024), 1514–1524. https://doi.org/10.1038/s41592-024-02272-z
- J. Abramson, J. Adler, J. Dunger, R. Evans, T. Green, A. Pritzel, O. Ronneberger, L. Willmore, A. J. Ballard, J. Bambrick, S. W. Bodenstein, D. A. Evans, C.-C. Hung, M. O'Neill, D. Reiman, K. Tunyasuvunakool, Z. Wu, A. Žemgulytė, E. Arvaniti, C. Beattie, O. Bertolli, A. Bridgland, A. Cherepanov, M. Congreve, A. I. Cowen-Rivers, A. Cowie, M. Figurnov, F. B. Fuchs, H. Gladman, R. Jain, Y. A. Khan, C. M. R. Low, K. Perlin, A. Potapenko, P. Savy, S. Singh, A. Stecula, A. Thillaisundaram, C. Tong, S. Yakneen, E. D. Zhong, M. Zielinski, A. Žídek, V. Bapst, P. Kohli, M. Jaderberg, D. Hassabis, and J. Jumper, “Accurate structure prediction of biomolecular interactions with AlphaFold 3,” Nature, vol. 630, no. 8016, pp. 493–500, 2024.
- Saro Passaro, Gabriele Corso, Jeremy Wohlwend, Mateo Reveiz, Stefan Thaler, Vignesh Ram Somnath, Noah Getz, Tally Portnoi, Julien Roy, Hannes Stark, Daniel Kwabi-Addo, Dominique Beaini, Tommi Jaakkola, and Regina Barzilay. 2025. Boltz-2: Towards accurate and efficient binding affinity prediction. bioRxiv. https://doi.org/10.1101/2025.06.14.659707
- Zeming Lin, Halil Akin, Roshan Rao, Brian Hie, Zhongkai Zhu, Wenting Lu, Nikita Smetanin, Robert Verkuil, Ori Kabeli, Yaniv Shmueli, Allan dos Santos Costa, Maryam Fazel-Zarandi, Tom Sercu, Sal Candido, and Alexander Rives. 2023. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 6637 (2023), 1123–1130. https://doi.org/10.1126/science.ade2574
- Alexander Rives, Joshua Meier, Tom Sercu, Siddharth Goyal, Zeming Lin, Jason Liu, Demi Guo, Myle Ott, C. Lawrence Zitnick, Jerry Ma, and Rob Fergus. 2021. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proceedings of the National Academy of Sciences 118, 15 (2021), e2016239118. https://doi.org/10.1073/pnas.2016239118
- Thomas Wolf, Lysandre Debut, Victor Sanh, Julien Chaumond, Clement Delangue, Anthony Moi, Pierric Cistac, Tim Rault, Remi Louf, Morgan Funtowicz, Joe Davison, Sam Shleifer, Patrick von Platen, Clara Ma, Yacine Jernite, Julien Plu, Canwen Xu, Teven Le Scao, Sylvain Gugger, Mariama Drame, Quentin Lhoest, and Alexander Rush. 2020. Transformers: State-of-the-Art Natural Language Processing. In Proceedings of the 2020 Conference on Empirical Methods in Natural Language Processing: System Demonstrations. ACL, 38–45. https://doi.org/10.18653/v1/2020.emnlp-demos.6
- AI2Science. 2026. VizFold Foundations: AlphaFold2/OpenFold2 interpretability and visualization, attention, representations, and intermediate protein structures. Retrieved March 2026 from https://github.com/AI2Science/vizfold-foundation
- Jeremy Wohlwend. 2025. Boltz [Computer software]. Retrieved March 2026 from https://github.com/jwohlwend/boltz
- Adam Paszke, Sam Gross, Francisco Massa, Adam Lerer, James Bradbury, Gregory Chanan, Trevor Killeen, Zeming Lin, Natalia Gimelshein, Luca Antiga, Alban Desmaison, Andreas Kopf, Edward Yang, Zachary DeVito, Martin Raison, Alykhan Tejani, Sasank Chilamkurthy, Benoit Steiner, Lu Fang, Junjie Bai, and Soumith Chintala. 2019. PyTorch: An Imperative Style, High-Performance Deep Learning Library. In Advances in Neural Information Processing Systems 32. 8024–8035.
- National Institute of Standards and Technology. 2025. CVE-2025-32434 detail. Retrieved March 2026 from https://nvd.nist.gov/vuln/detail/CVE-2025-32434
- RDKit. 2025. rdkit.rdBase module documentation. Retrieved March 2026 from https://www.rdkit.org/docs/source/rdkit.rdBase.html

This work is licensed under a Creative Commons Attribution 4.0 International License.
PEARC '26, Minneapolis, MN, USA
© 2026 Copyright held by the owner/author(s).
ACM ISBN 979-8-4007-2377-3/26/07.
DOI: https://doi.org/10.1145/3785462.3815886